
Accueil » Patients et proches » Le rétinoblastome
Vous voulez approfondir vos connaissances sur les cancers de l’enfant, les traitements, les conseils concernant la vie à l’hôpital, à la maison, la réadaptation, et toutes les situations que l’on peut rencontrer.
Les réponses à vos questions
Vous pouvez trouver la totalité de cet article sur le site de l’Institut National du Cancer dédié aux cancers de l’enfant.

Le rétinoblastome est une tumeur cancéreuse rare de la rétine se manifestant généralement avant l’âge de 5 ans. Il s’agit d’une maladie d’origine souvent génétique qui a un excellent taux de guérison en France.
Le rétinoblastome est une tumeur cancéreuse de l’œil qui atteint les cellules de la rétine. C’est un cancer rare qui touche environ 50 enfants par an en France, essentiellement les nourrissons et les jeunes enfants avant l’âge de 5 ans. Le rétinoblastome est un cancer dont on guérit s’il est détecté de façon précoce (95% de taux de guérison dans les pays développés, voire près de 100% en France).
La rétine est une fine membrane nerveuse qui tapisse l’intérieur de l’œil. Elle contient les cellules photoréceptrices qui captent les rayons lumineux pour les transmettre au nerf optique jusqu’au système nerveux central. Celui-ci analyse l’information et la traduit en image.
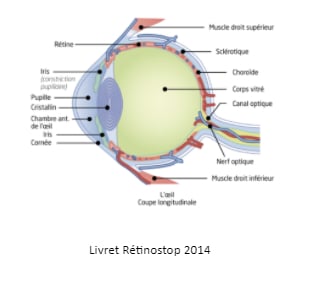
Le rétinoblastome est une maladie d’origine fréquemment génétique. Il peut se manifester dans un seul œil, on parle alors de rétinoblastome unilatéral (60% des cas) ou dans les deux yeux, on parle alors rétinoblastome bilatéral (40% des cas).
Le rétinoblastome est causé par des changements (mutations) subis par le gène 1 du rétinoblastome (RB1). RB1 est l’un des gènes responsables du contrôle de la division cellulaire. Chez les individus porteurs d’une mutation du gène RB1, la division cellulaire, non régulée, entraîne la survenue des tumeurs rétiniennes. Il y a 2 copies du gène RB1 dans chaque cellule. Le changement doit affecter les deux copies du gène RB1 dans une cellule de la rétine en développement pour qu’un rétinoblastome apparaisse.
Dans un peu plus de la moitié des cas, le rétinoblastome n’est pas héréditaire. C’est-à-dire que l’enfant n’a pas hérité d’une mutation génétique mais celle-ci est survenue au hasard après la conception (union du spermatozoïde et de l’ovule); dans ce cas, les mutations ne surviennent qu’au niveau de la rétine.
Dans les cas de rétinoblastome héréditaire, la mutation est soit transmise à l’enfant par l’un des parents qui est porteur de la mutation du gène RB1, soit survenue dans une des cellules aboutissant à la formation du spermatozoïde ou de ovule avant la conception. Chez les enfants atteints de formes héréditaires, le rétinoblastome se développe généralement dans les deux yeux (forme bilatérale). Tous ces enfants sont susceptibles de transmettre la mutation RB1 à leurs propres enfants. Ils risquent également davantage d’avoir d’autres cancers et nécessitent donc un suivi médical approprié.
Lorsque la mutation du gène RB1 est identifiée chez une personne ayant eu un rétinoblastome, des tests individuels sont proposés aux membres de la famille potentiellement porteurs de la mutation. Cela a pour but de mettre en place une surveillance ophtalmologique pour dépister précocement l’apparition possible de la maladie chez les enfants. Toutes les familles se voient donc proposer une consultation d’information génétique.
Les signes cliniques caractéristiques sont :
En cas de doute, vous pouvez faire le test suivant : placez votre enfant dans une pièce un peu sombre et prenez une photo avec le flash sans le système anti yeux rouges. Normalement, la pupille est orangée. Si elle est blanche, c’est un signe pour consulter un ophtalmologiste.
D’autres signes cliniques plus rares peuvent se manifester telle que l’augmentation de volume de l’œil pouvant devenir douloureux avec rougeur péri oculaire (buphtalmie).
Le diagnostic est un processus qui permet d’identifier la cause d’un problème de santé. Il débute habituellement par une visite à votre médecin. Si nécessaire, ce dernier dirigera votre enfant vers un spécialiste ou lui prescrira des examens complémentaires.
Le diagnostic du rétinoblastome repose essentiellement sur l’examen du fond d’œil sous anesthésie générale. Il précise le nombre de tumeurs, leur taille et leur localisation. Une échographie oculaire peut parfois être utile pour préciser le diagnostic, mais elle ne remplacera pas le fond d’oeil.
Une IRM permettra de vérifier que le nerf optique lui-même, en arrière de l’oeil, n’est pas atteint.
Les traitements sont efficaces, permettant de guérir de près de 100% des enfants en France. L’objectif du traitement est de guérir l’enfant en préservant, autant que possible, l’œil et la vision.
Le traitement sera adapté au cas de l’enfant et de sa maladie :
Dans les pays industrialisés, l’immense majorité des patients atteints de rétinoblastome guérit. L’enjeu de la recherche est donc essentiellement de diminuer les séquelles des traitements, en particulier pour préserver au mieux la vision de l’enfant. Un suivi adapté du fond d’œil après les traitements est mis en place dans tous les cas. Il permet de détecter à temps une éventuelle récidive et la traiter le plus vite possible pour conserver la vision de l’œil atteint. Le suivi pédiatrique spécialisé est également important pour détecter des séquelles du traitement pas chimiothérapie et pour transmettre à l’enfant devenu jeune adulte les recommandations dont il peut avoir besoin pour sa descendance.
A télécharger :
Livret d’information sur le rétinoblastome de l’Institut Curie et de l’association Rétinostop : https://retinostop.org/documents/livret-retinoblastome-curie-retinostop-2014.pdf
Copyright © 2021 SFCE – Theme by IntegDevelop – Tous droits réservés